

引用本文: 李廣麗, 王中鳴, 李振光, 盧軍, 彭瓊. 四例Dyke-Davidoff-Masson綜合征癲癇發作及視頻腦電圖特點. 癲癇雜志, 2023, 9(6): 466-471. doi: 10.7507/2096-0247.202309001 復制
Dyke-Davidoff-Masson綜合征(Dyke-Davidoff-Masson syndrome,DDMS)是一種罕見的神經發育異常綜合征。它主要特征是一側大腦半球在發育或萎縮方面存在不同程度的異常,同時伴隨著顱骨的代償性改變[1]。Dyke、Davidoff和Masson于1933年首次報道了該病,DDMS在臨床上可能表現為偏癱、智力發育遲緩、抽搐、語言障礙和面部不對稱等癥狀[2]。近年來,隨著科學和經濟的發展,電子計算機斷層掃描(Computed tomography,CT)和磁共振成像(Magnetic resonance imaging,MRI)等醫學影像技術在臨床應用中得到了廣泛的應用。這也導致了與DDMS相關的病例報道逐漸增多。目前國內外相關病例報道僅百余例[3]。本文總結我院確診的4例DDMS病例臨床表現(尤其是癲癇發作)及視頻腦電圖(Video-electroencephalogram,VEEG)癇樣放電特點等資料并進行相關文獻復習,探討DDMS的癲癇發作特點、VEEG放電情況、治療及效果,以期為臨床診治DDMS提供一些參考。
病例資料 患者1 女,19歲,因“右側肢體抽搐伴疼痛3個月余,加重10天”入院。自備抗癲癇發作藥物左乙拉西坦 0.5 g口服,每日兩次。患者足月出生,無難產史,無明顯外傷史,其父母非近親結婚,家族中無特殊、類似及遺傳病史可詢。家屬代訴2歲時“流行性乙型腦炎”病史,遺留右側肢體活動不利,從小學開始學習困難,記憶力與理解力較同齡人稍差,中專在讀。體格檢查: 面部對稱,左側肢體肌力、肌張力正常。右側上、下肢近端肌力均為4級,遠端肌力均為3級,右側肌肉稍萎縮,肌張力正常,呈右側輕偏癱步態。住院期間右側肢體間斷抽搐,每天發作數次,持續約30 s。診斷:癥狀性癲癇,局灶起始伴知覺保留肌陣攣發作。
患者2 女,14歲,因“腦外傷后反復發作性意識喪失伴肢體抽搐13年余”入院。自備抗癲癇發作藥物奧卡西平片0.6 g、左乙拉西坦片1.0 g 、托吡酯片 25 mg ,均為口服,每日兩次。患者足月出生,無難產史,其父母非近親結婚,家族中無特殊、類似及遺傳病史可詢。家屬代訴1歲時有“頭部外傷史”,自此智力低下,學習困難,小學學歷。體格檢查:面部不對稱,左側鼻唇溝變淺,右側肢體肌力、肌張力正常,左上肢近端肌力3級,遠端肌力2級,手指精細運動差。左下肢近端肌力4級,遠端肌力2級,左側肌肉稍萎縮,左側肢體肌張力正常,呈左側偏癱步態。診斷:癥狀性癲癇,全面性起始伴運動癥狀強直-陣攣發作;腦外傷后遺癥。
患者3 女,24歲,因“反復發作性意識障礙伴肢體抽搐22年余,伴精神行為異常3個月”入院。自備抗癲癇發作藥物左乙拉西坦片0.5 g口服,每日兩次。患者足月出生,無難產史,其父母非近親結婚,家族中無特殊、類似及遺傳病史可詢。家屬代訴2歲“腦外傷史”,自幼學習能力差,智力偏低,反應遲鈍。體格檢查:面部對稱,左側上肢近端肌力4級,遠端肌力3級,肌張力增高,長期保持于胸前,呈爪型手姿勢,無法行手指精細運動。左下肢肌力、肌張力正常,右側肢體肌力、肌張力正常。步態正常。診斷:癥狀性癲癇,局灶起始伴知覺障礙強直進展為全面性強直-陣攣發作;腦外傷后遺癥。
患者4 女,6歲,因“間斷抽搐17個月余”入院。自備抗癲癇發作藥物左乙拉西片0.25 g、奧卡西平片 0.3 g,均為口服,每日兩次。患者足月出生,無難產史,其父母非近親結婚,既往“新生兒腦損傷”病史。3歲11月齡時有1次“熱性驚厥”病史。查體:面部對稱,右上肢近端肌力4級,遠端肌力3級,手指精細運動差,右下肢肌力5-級,右足內翻畸形,肌張力稍增高,肌肉萎縮。左側肢體肌力、肌張力正常,呈右側偏癱步態。診斷:癥狀性癲癇,局灶起始伴知覺障礙強直進展為全面性強直-陣攣發作;左側顳、頂葉腦軟化灶。4例患者顱腦影像學及腦電圖表現見表1和圖1、2、3。
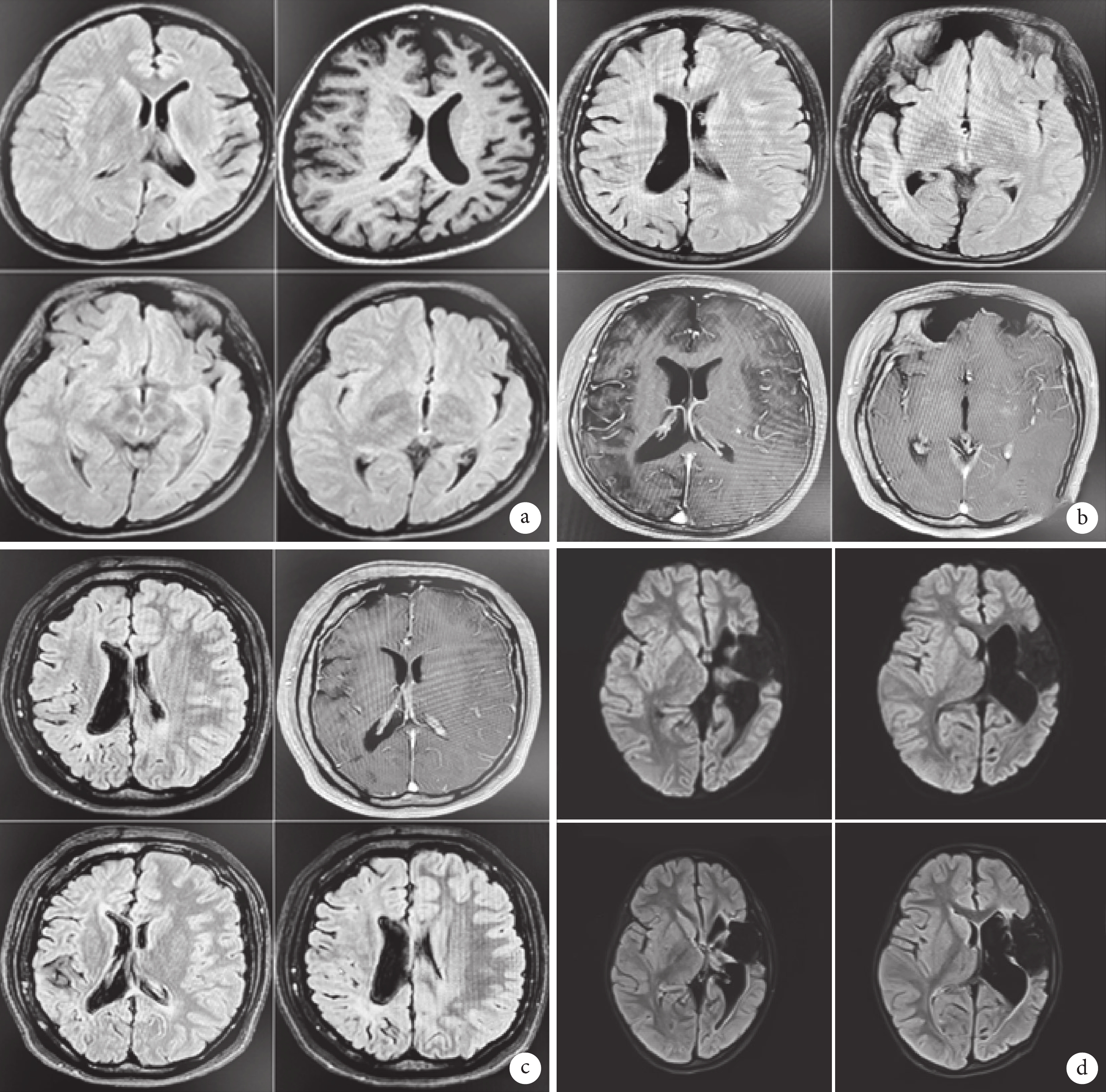
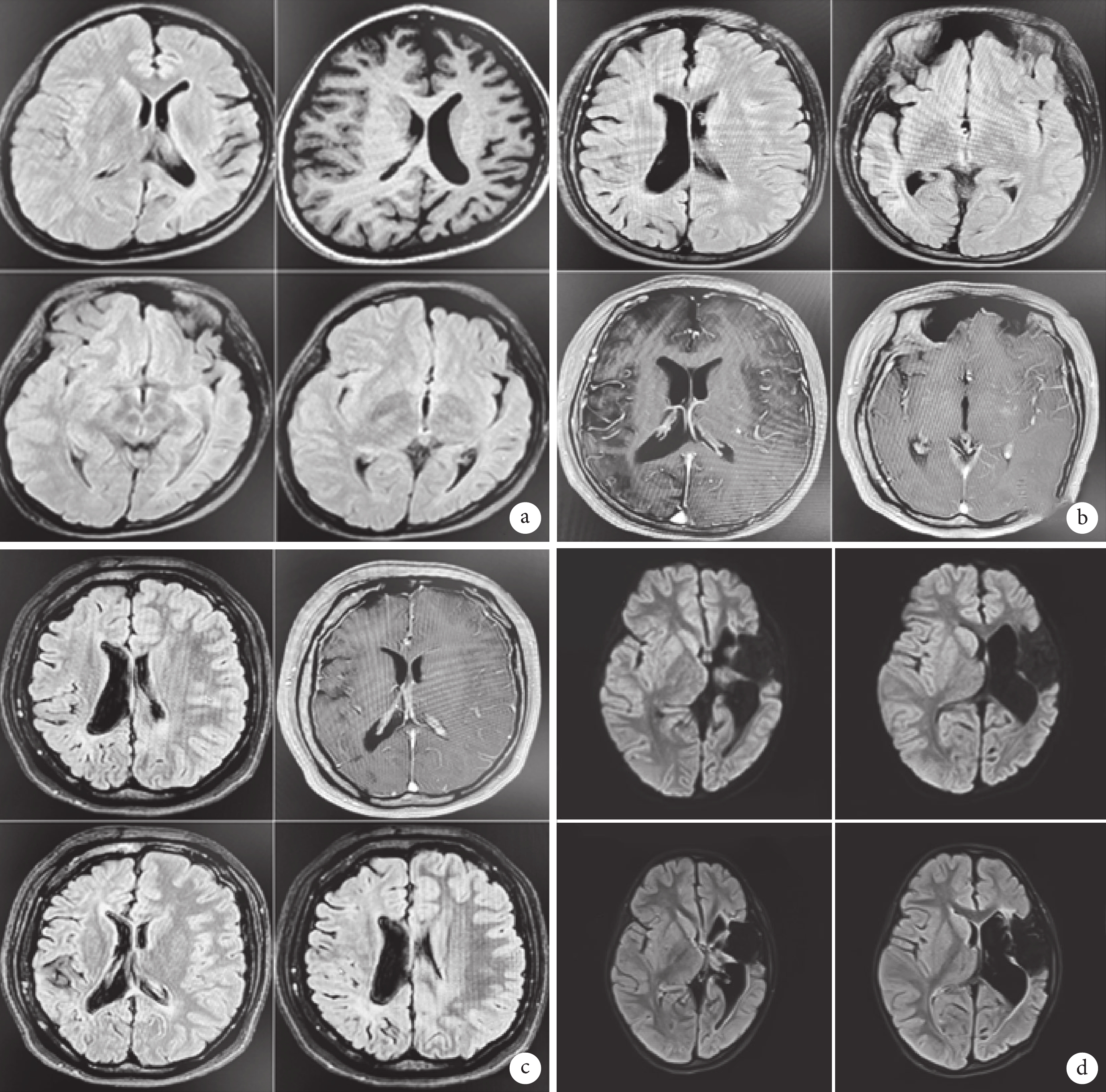 圖1
4例患者的頭顱MRI結果
圖1
4例患者的頭顱MRI結果
a. 患者1左側大腦萎縮、左側顱骨略增厚、左側額竇擴大;b. 患者2右側大腦半球體積縮小腦溝增寬,右側腦室擴大,右側額、顳、頂部顱骨增厚,右側額竇擴大;c. 患者3右側大腦半球體積縮小,伴右側顱骨增厚及右側蝶竇、額竇擴大;d. 患者4 左側大腦半球萎縮,左側腦室擴大、左側額竇擴大,左側顱骨略增厚,左側額、顳島葉軟化灶
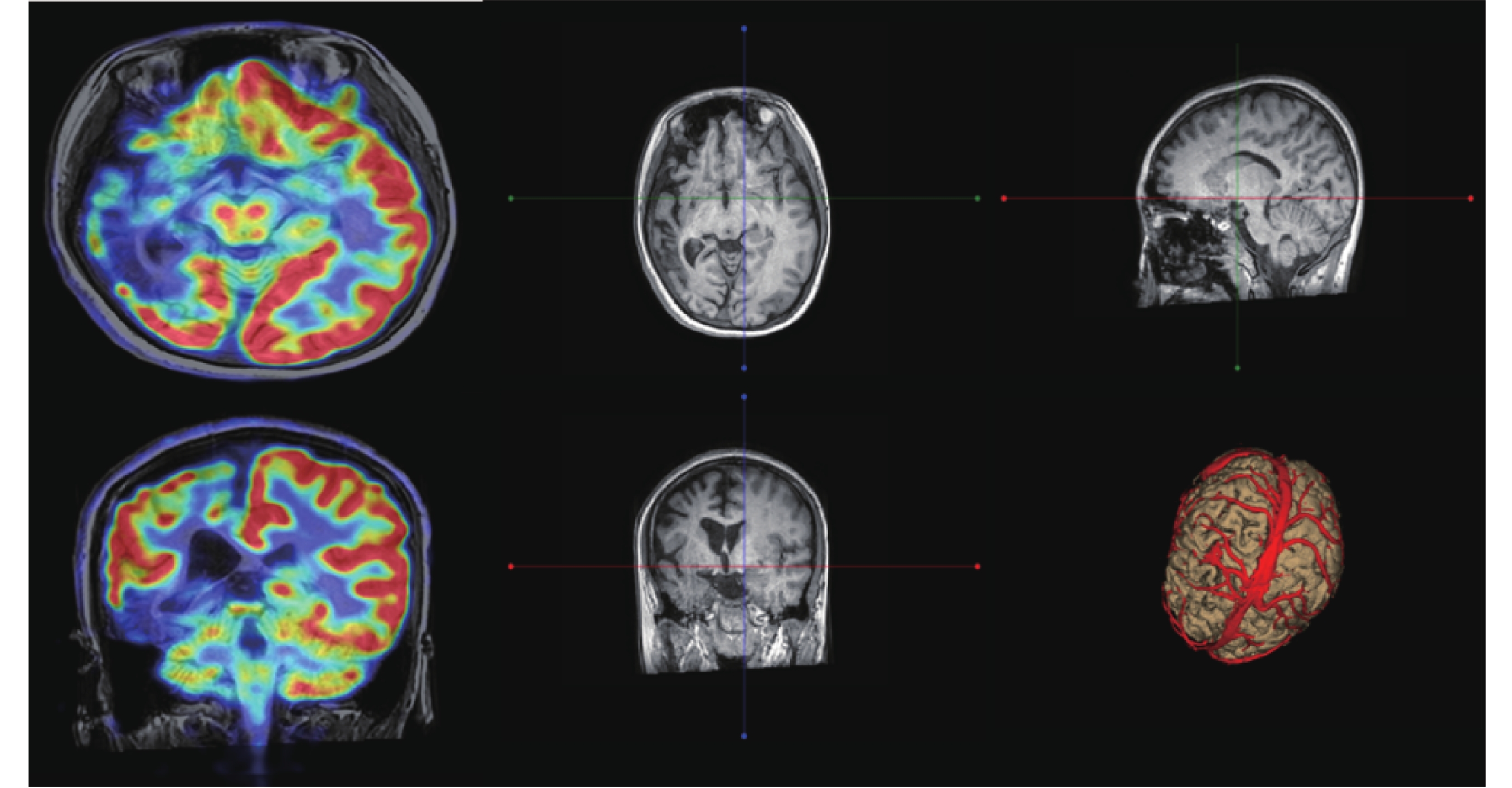
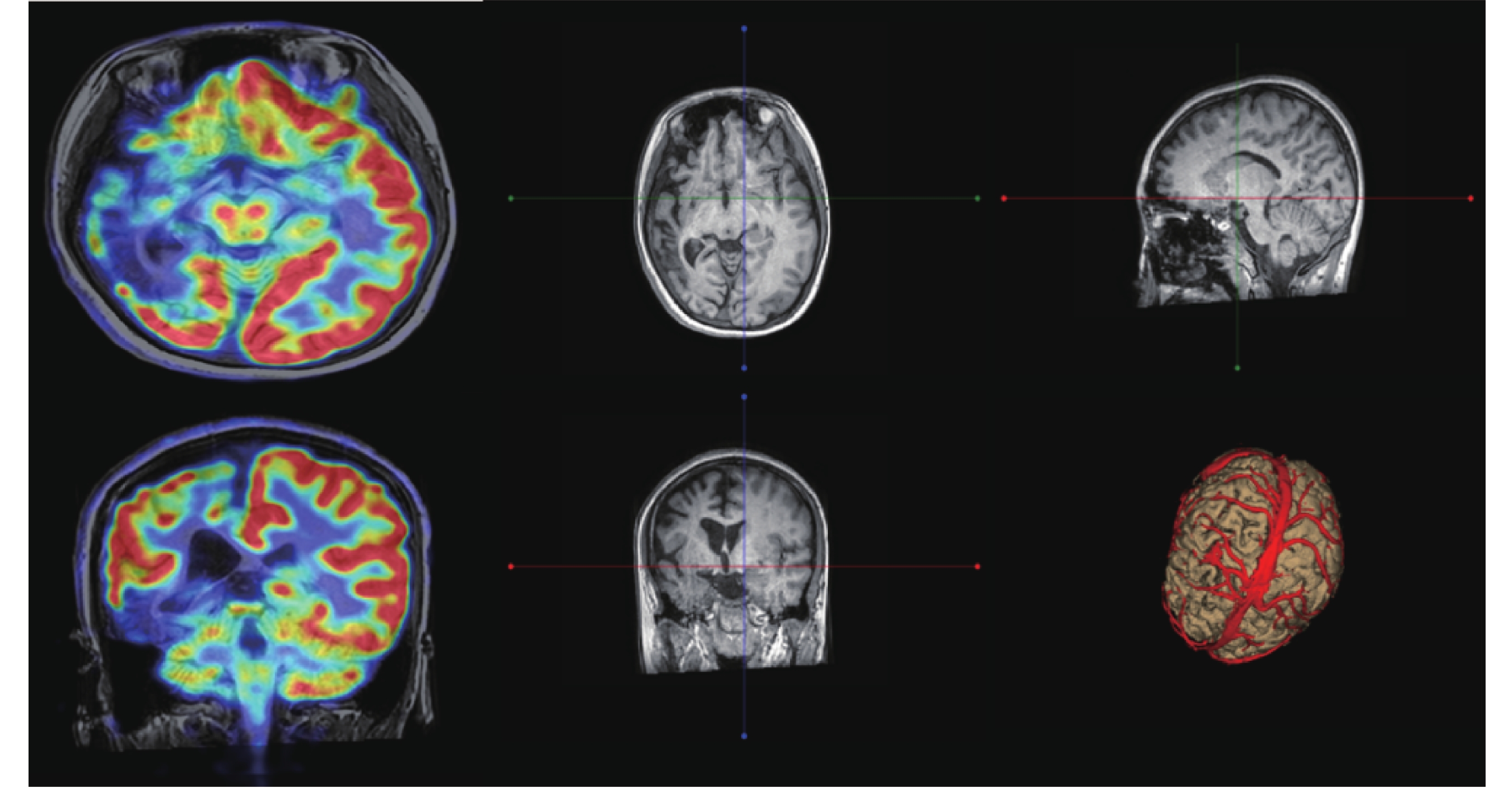 圖2
患者2的PET-CT及MRI(癲癇序列)
圖2
患者2的PET-CT及MRI(癲癇序列)
腦部PET-CT:右側額顳頂枕葉放射性分布較左側減低,右側腦萎縮,頭頸部其他部位未見明顯異常
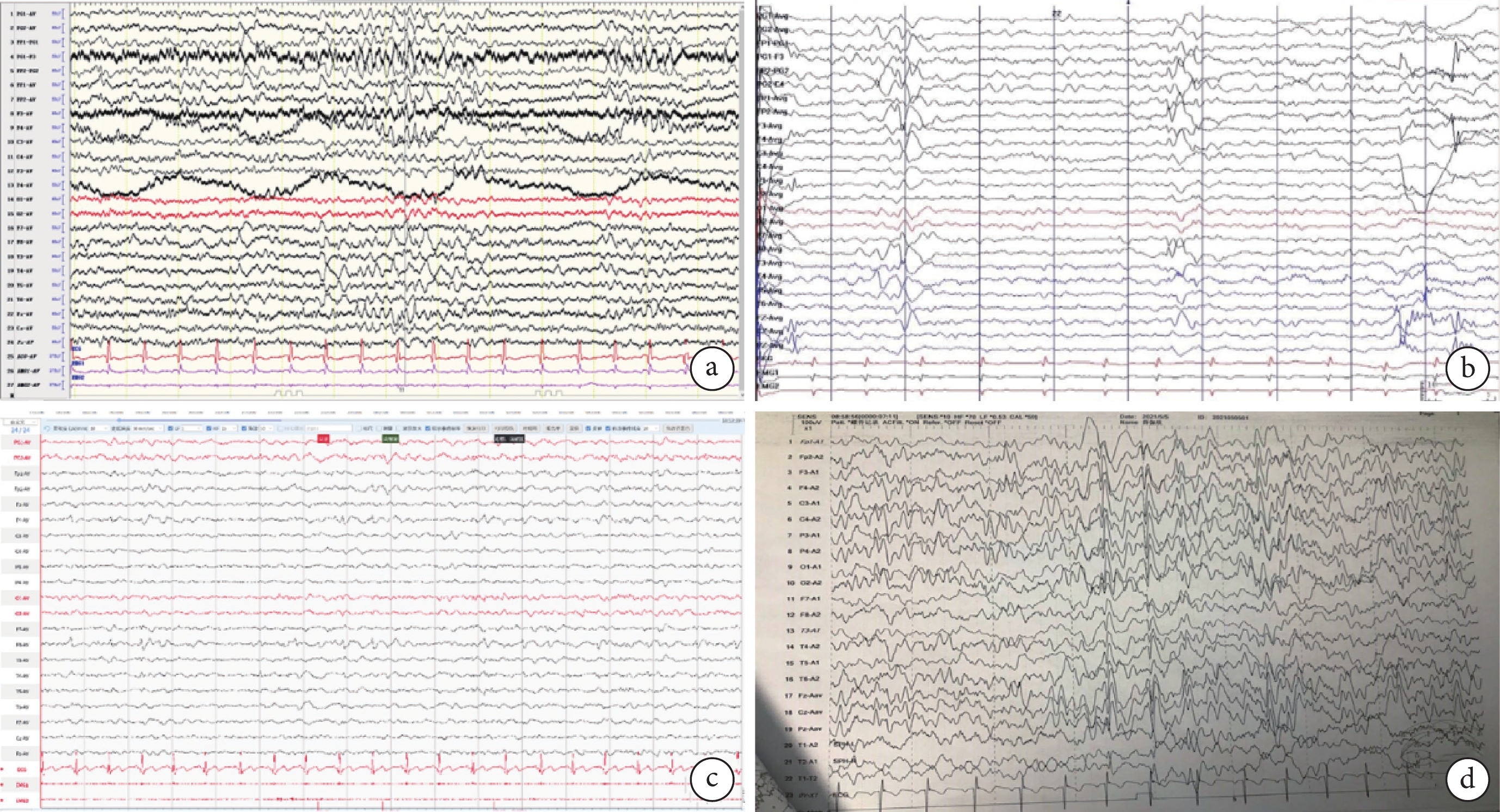
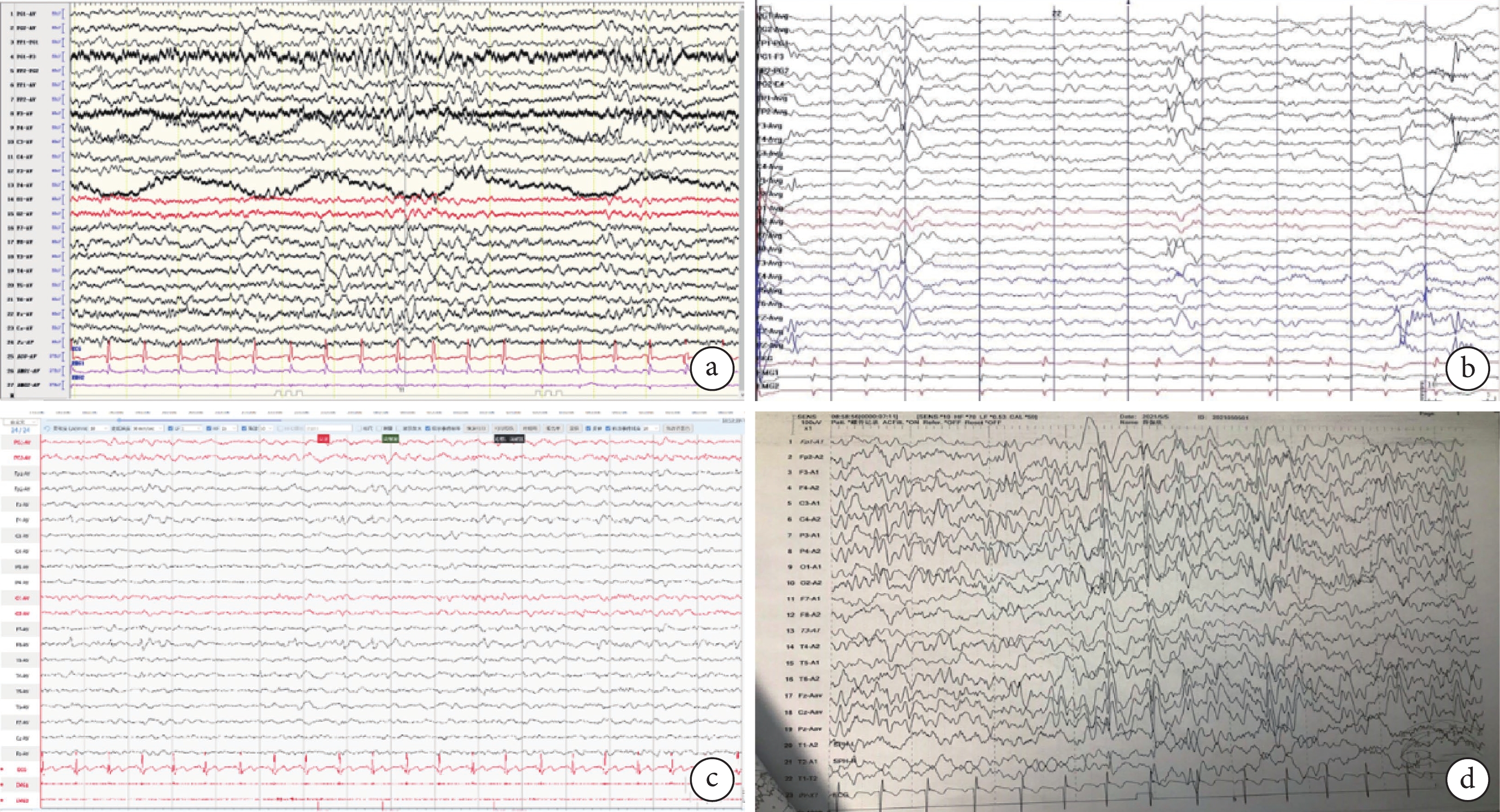 圖3
4例患者的視頻腦電圖
圖3
4例患者的視頻腦電圖
a. 患者1左側額、顳區慢波活動分布增多;雙側額中央、前顳區(左側為甚),尖慢復合波發放;b. 患者2 雙側額極、額區、前顳區尖、棘慢復合波放電(右側為著),右側蝶骨電極、前顳、中顳尖波、尖慢波放電;c. 患者3右側額、中央及前顳區為著中-極高等波幅慢波活動夾雜尖波、尖慢復合波呈短-中程頻繁發放;d. 患者4左側額中央、頂前顳區尖樣慢波活動夾雜尖波、尖慢復合波短-中程發放
討論 DDMS是一種罕見的中樞神經系統疾病,其診斷是基于影像學和臨床表現的綜合診斷。其影像學典型表現為:單側大腦半球萎縮、同側側腦室擴張、同側顱骨增生、鼻竇及乳突竇增大[4]。本文總結的4例DDMS患者均符合該影像學表現。其臨床表現包括難治性癲癇、對側偏癱或無力、智力低下及面部不對稱,智力低下和面部不對稱等表現通常在腦萎縮或明顯發育不良的患者中更為常見。少數患者出現感覺異常、言語障礙、認知障礙和情感不穩定等癥狀。在極少數情況下,DDMS可能與甲狀腺功能減退或腎上腺功能減退等內分泌系統問題相關聯[5]。本文4例DDMS患者臨床表現均為癲癇發作、智力低下、學習困難,3例患者存在偏癱步態,1例患者面部不對稱,其中1例患者精神行為異常。
DDMS病因眾多,研究者通常將其分為原發型和繼發型[6]。原發型可能是由胎兒期或嬰兒期宮腔內血管損傷引起的。這種損傷可能是由于腦動脈異常或缺血性事件導致的血供不足,從而影響了腦半球的正常發育。繼發型可能是由其他原因引起的,如腦外傷、感染、腦血管疾病或神經退行性疾病等。這些因素導致的DDMS在病因和臨床表現上可能與原發型有所不同 [7]。本文4例DDMS患者均為女性,患者1既往“流行性乙型腦炎”病史,患者2和3既往“腦外傷”病史,患者4既往“新生兒腦損傷、熱性驚厥”等病史,病因學上均考慮繼發性DDMS。
癲癇是一種慢性腦部疾病,具有不同的病因基礎和臨床表現,但其共同特征是反復發作的癲癇發作。其病因學大致可以歸納為以下幾個方面:結構性、代謝性、免疫性、感染性以及病因不明。這些因素都可能導致腦部功能異常,從而引發癲癇發作的發生[8]。本文4例DDMS患者均診斷為癥狀性癲癇,從病因學上屬于結構性病變。
癲癇的診斷不僅僅依據患者典型的臨床表現,更重要的是通過腦電圖監測。腦電圖癇樣放電被認為是目前診斷癲癇的金標準。根據國際抗癲癇聯盟的定義,癲癇發作的分類基于腦電圖放電的起源部位,即放電是起源于一側大腦半球還是同時涉及雙側大腦皮質的致癇網絡。根據這個分類,癲癇發作可以分為局灶性發作和全面性發作[9]。因此,腦電圖癇樣波放電范圍與癲癇發作特點是密不可分的。本文4例DDMS患者均存在典型的癇樣放電。患者1 VEEG顯示雙側額中央、前顳區(左側為甚)癇樣放電,癇樣放電范圍雙側大腦皮質額中央,左側前顳區為主,其癲癇發作特點是局灶性發作,右側肢體抽搐伴疼痛,考慮未采集發作間期的VEEG,無法判斷癲癇放電從哪里起源,高度懷疑起源于左側大腦皮質致癇網絡區。患者2 VEEG顯示雙側額極、額區、前顳區可見癇樣放電,右側蝶骨電極、前顳、中顳區也可見癇樣放電,且以右側為主。其癲癇發作特點是全面性發作,意識喪失伴四肢抽搐,右側肢體抽搐更為厲害。VEEG癇樣放電范圍與患者臨床癲癇發作基本一致。患者3 VEEG顯示右側額、中央及前顳區癇樣放電,癇樣放電僅局限于右側額、中央、前顳區,其癲癇發作特點意識喪失、左側肢體抽搐,繼而全身抽搐,考慮采集VEEG時患者未出現癲癇發作,高度懷疑發作期VEEG癇樣放電由右側大腦半球傳導至對側而出現的臨床表現。患者4 VEEG顯示左側額中央、頂前顳區癲樣放電,癇樣放電僅局限于左側額中央、頂前顳區,其癲癇發作特點右側肢體陣攣,伴或不伴左側肢體陣攣,考慮VEEG采集時患者未出現癲癇發作,高度懷疑發作期VEEG癇樣放電由左側大腦半球傳導至對側而出現的臨床表現。
目前,控制DDMS癲癇發作的治療方案主要包括藥物治療、手術治療、神經調控和生酮飲食等。藥物治療是最常見和常規的治療方式,通過使用抗癲癇發作藥物來減少癲癇發作的頻率和嚴重程度。如果藥物治療無效或存在嚴重的副作用,手術治療可以考慮[10]。本文4例DDMS患者的抗癲癇發作治療方案:患者1、患者3及患者4以聯合多種抗癲癇發作藥物控制癲癇發作,患者1發作間期VEEG大腦皮質雙側均有癇樣放電,臨床發作特點屬于頑固性局灶性發作,住院期間首先選用拉莫三嗪片無法控制右側肢體抽搐癥狀,后口服加用吡侖帕奈片也不能完全控制癥狀,仍有輕微右上肢抽搐癥狀,最后苯巴比妥注射液每12 h一次,完全控制癥狀。出院后停用苯巴比妥注射液,加用苯巴比妥片。患者2屬于難治性癲癇,1歲左右出現癲癇發作,期間反復調整抗癲癇藥物及聯合3~4種抗癲癇發作藥物仍不能控制癲癇發作,基本每天出現癲癇發作數次,最后予以外科手術治療行大腦半球離斷術,術后繼續口服奧卡西平片、左乙拉西坦片、拉考沙胺片等抗癲癇藥物治療。患者3發作間期VEEG癇樣放電局限于右側大腦半球皮質,入院前口服奧卡西平不能完全控制癲癇發作癥狀,后加用左乙拉西坦片可控制癲癇發作。患者4既往新生兒腦損傷、熱性驚厥等病史,本次發作間期VEEG癇樣放電局限于左側大腦半球皮質,首先使用左乙拉西坦不能控制癲癇發作,后加用奧卡西平控制癲癇發作。以上4例DDMS患者住院期間調整抗癲癇發作治療方案后,出院隨訪仍有間斷發作情況,總體而言癲癇發作控制較滿意。
DDMS是一種罕見的腦部綜合征,會導致癲癇發作。治療的主要目標是控制癲癇發作。為了提高疾病預后,癲癇專科醫生應該增強對DDMS的認識,提高該疾病的診斷率,并在早期發現和早期診斷方面做好工作,以便進行合理的治療。
利益沖突聲明 所有作者無利益沖突。
Dyke-Davidoff-Masson綜合征(Dyke-Davidoff-Masson syndrome,DDMS)是一種罕見的神經發育異常綜合征。它主要特征是一側大腦半球在發育或萎縮方面存在不同程度的異常,同時伴隨著顱骨的代償性改變[1]。Dyke、Davidoff和Masson于1933年首次報道了該病,DDMS在臨床上可能表現為偏癱、智力發育遲緩、抽搐、語言障礙和面部不對稱等癥狀[2]。近年來,隨著科學和經濟的發展,電子計算機斷層掃描(Computed tomography,CT)和磁共振成像(Magnetic resonance imaging,MRI)等醫學影像技術在臨床應用中得到了廣泛的應用。這也導致了與DDMS相關的病例報道逐漸增多。目前國內外相關病例報道僅百余例[3]。本文總結我院確診的4例DDMS病例臨床表現(尤其是癲癇發作)及視頻腦電圖(Video-electroencephalogram,VEEG)癇樣放電特點等資料并進行相關文獻復習,探討DDMS的癲癇發作特點、VEEG放電情況、治療及效果,以期為臨床診治DDMS提供一些參考。
病例資料 患者1 女,19歲,因“右側肢體抽搐伴疼痛3個月余,加重10天”入院。自備抗癲癇發作藥物左乙拉西坦 0.5 g口服,每日兩次。患者足月出生,無難產史,無明顯外傷史,其父母非近親結婚,家族中無特殊、類似及遺傳病史可詢。家屬代訴2歲時“流行性乙型腦炎”病史,遺留右側肢體活動不利,從小學開始學習困難,記憶力與理解力較同齡人稍差,中專在讀。體格檢查: 面部對稱,左側肢體肌力、肌張力正常。右側上、下肢近端肌力均為4級,遠端肌力均為3級,右側肌肉稍萎縮,肌張力正常,呈右側輕偏癱步態。住院期間右側肢體間斷抽搐,每天發作數次,持續約30 s。診斷:癥狀性癲癇,局灶起始伴知覺保留肌陣攣發作。
患者2 女,14歲,因“腦外傷后反復發作性意識喪失伴肢體抽搐13年余”入院。自備抗癲癇發作藥物奧卡西平片0.6 g、左乙拉西坦片1.0 g 、托吡酯片 25 mg ,均為口服,每日兩次。患者足月出生,無難產史,其父母非近親結婚,家族中無特殊、類似及遺傳病史可詢。家屬代訴1歲時有“頭部外傷史”,自此智力低下,學習困難,小學學歷。體格檢查:面部不對稱,左側鼻唇溝變淺,右側肢體肌力、肌張力正常,左上肢近端肌力3級,遠端肌力2級,手指精細運動差。左下肢近端肌力4級,遠端肌力2級,左側肌肉稍萎縮,左側肢體肌張力正常,呈左側偏癱步態。診斷:癥狀性癲癇,全面性起始伴運動癥狀強直-陣攣發作;腦外傷后遺癥。
患者3 女,24歲,因“反復發作性意識障礙伴肢體抽搐22年余,伴精神行為異常3個月”入院。自備抗癲癇發作藥物左乙拉西坦片0.5 g口服,每日兩次。患者足月出生,無難產史,其父母非近親結婚,家族中無特殊、類似及遺傳病史可詢。家屬代訴2歲“腦外傷史”,自幼學習能力差,智力偏低,反應遲鈍。體格檢查:面部對稱,左側上肢近端肌力4級,遠端肌力3級,肌張力增高,長期保持于胸前,呈爪型手姿勢,無法行手指精細運動。左下肢肌力、肌張力正常,右側肢體肌力、肌張力正常。步態正常。診斷:癥狀性癲癇,局灶起始伴知覺障礙強直進展為全面性強直-陣攣發作;腦外傷后遺癥。
患者4 女,6歲,因“間斷抽搐17個月余”入院。自備抗癲癇發作藥物左乙拉西片0.25 g、奧卡西平片 0.3 g,均為口服,每日兩次。患者足月出生,無難產史,其父母非近親結婚,既往“新生兒腦損傷”病史。3歲11月齡時有1次“熱性驚厥”病史。查體:面部對稱,右上肢近端肌力4級,遠端肌力3級,手指精細運動差,右下肢肌力5-級,右足內翻畸形,肌張力稍增高,肌肉萎縮。左側肢體肌力、肌張力正常,呈右側偏癱步態。診斷:癥狀性癲癇,局灶起始伴知覺障礙強直進展為全面性強直-陣攣發作;左側顳、頂葉腦軟化灶。4例患者顱腦影像學及腦電圖表現見表1和圖1、2、3。
圖1
4例患者的頭顱MRI結果
a. 患者1左側大腦萎縮、左側顱骨略增厚、左側額竇擴大;b. 患者2右側大腦半球體積縮小腦溝增寬,右側腦室擴大,右側額、顳、頂部顱骨增厚,右側額竇擴大;c. 患者3右側大腦半球體積縮小,伴右側顱骨增厚及右側蝶竇、額竇擴大;d. 患者4 左側大腦半球萎縮,左側腦室擴大、左側額竇擴大,左側顱骨略增厚,左側額、顳島葉軟化灶
圖2
患者2的PET-CT及MRI(癲癇序列)
腦部PET-CT:右側額顳頂枕葉放射性分布較左側減低,右側腦萎縮,頭頸部其他部位未見明顯異常
圖3
4例患者的視頻腦電圖
a. 患者1左側額、顳區慢波活動分布增多;雙側額中央、前顳區(左側為甚),尖慢復合波發放;b. 患者2 雙側額極、額區、前顳區尖、棘慢復合波放電(右側為著),右側蝶骨電極、前顳、中顳尖波、尖慢波放電;c. 患者3右側額、中央及前顳區為著中-極高等波幅慢波活動夾雜尖波、尖慢復合波呈短-中程頻繁發放;d. 患者4左側額中央、頂前顳區尖樣慢波活動夾雜尖波、尖慢復合波短-中程發放
討論 DDMS是一種罕見的中樞神經系統疾病,其診斷是基于影像學和臨床表現的綜合診斷。其影像學典型表現為:單側大腦半球萎縮、同側側腦室擴張、同側顱骨增生、鼻竇及乳突竇增大[4]。本文總結的4例DDMS患者均符合該影像學表現。其臨床表現包括難治性癲癇、對側偏癱或無力、智力低下及面部不對稱,智力低下和面部不對稱等表現通常在腦萎縮或明顯發育不良的患者中更為常見。少數患者出現感覺異常、言語障礙、認知障礙和情感不穩定等癥狀。在極少數情況下,DDMS可能與甲狀腺功能減退或腎上腺功能減退等內分泌系統問題相關聯[5]。本文4例DDMS患者臨床表現均為癲癇發作、智力低下、學習困難,3例患者存在偏癱步態,1例患者面部不對稱,其中1例患者精神行為異常。
DDMS病因眾多,研究者通常將其分為原發型和繼發型[6]。原發型可能是由胎兒期或嬰兒期宮腔內血管損傷引起的。這種損傷可能是由于腦動脈異常或缺血性事件導致的血供不足,從而影響了腦半球的正常發育。繼發型可能是由其他原因引起的,如腦外傷、感染、腦血管疾病或神經退行性疾病等。這些因素導致的DDMS在病因和臨床表現上可能與原發型有所不同 [7]。本文4例DDMS患者均為女性,患者1既往“流行性乙型腦炎”病史,患者2和3既往“腦外傷”病史,患者4既往“新生兒腦損傷、熱性驚厥”等病史,病因學上均考慮繼發性DDMS。
癲癇是一種慢性腦部疾病,具有不同的病因基礎和臨床表現,但其共同特征是反復發作的癲癇發作。其病因學大致可以歸納為以下幾個方面:結構性、代謝性、免疫性、感染性以及病因不明。這些因素都可能導致腦部功能異常,從而引發癲癇發作的發生[8]。本文4例DDMS患者均診斷為癥狀性癲癇,從病因學上屬于結構性病變。
癲癇的診斷不僅僅依據患者典型的臨床表現,更重要的是通過腦電圖監測。腦電圖癇樣放電被認為是目前診斷癲癇的金標準。根據國際抗癲癇聯盟的定義,癲癇發作的分類基于腦電圖放電的起源部位,即放電是起源于一側大腦半球還是同時涉及雙側大腦皮質的致癇網絡。根據這個分類,癲癇發作可以分為局灶性發作和全面性發作[9]。因此,腦電圖癇樣波放電范圍與癲癇發作特點是密不可分的。本文4例DDMS患者均存在典型的癇樣放電。患者1 VEEG顯示雙側額中央、前顳區(左側為甚)癇樣放電,癇樣放電范圍雙側大腦皮質額中央,左側前顳區為主,其癲癇發作特點是局灶性發作,右側肢體抽搐伴疼痛,考慮未采集發作間期的VEEG,無法判斷癲癇放電從哪里起源,高度懷疑起源于左側大腦皮質致癇網絡區。患者2 VEEG顯示雙側額極、額區、前顳區可見癇樣放電,右側蝶骨電極、前顳、中顳區也可見癇樣放電,且以右側為主。其癲癇發作特點是全面性發作,意識喪失伴四肢抽搐,右側肢體抽搐更為厲害。VEEG癇樣放電范圍與患者臨床癲癇發作基本一致。患者3 VEEG顯示右側額、中央及前顳區癇樣放電,癇樣放電僅局限于右側額、中央、前顳區,其癲癇發作特點意識喪失、左側肢體抽搐,繼而全身抽搐,考慮采集VEEG時患者未出現癲癇發作,高度懷疑發作期VEEG癇樣放電由右側大腦半球傳導至對側而出現的臨床表現。患者4 VEEG顯示左側額中央、頂前顳區癲樣放電,癇樣放電僅局限于左側額中央、頂前顳區,其癲癇發作特點右側肢體陣攣,伴或不伴左側肢體陣攣,考慮VEEG采集時患者未出現癲癇發作,高度懷疑發作期VEEG癇樣放電由左側大腦半球傳導至對側而出現的臨床表現。
目前,控制DDMS癲癇發作的治療方案主要包括藥物治療、手術治療、神經調控和生酮飲食等。藥物治療是最常見和常規的治療方式,通過使用抗癲癇發作藥物來減少癲癇發作的頻率和嚴重程度。如果藥物治療無效或存在嚴重的副作用,手術治療可以考慮[10]。本文4例DDMS患者的抗癲癇發作治療方案:患者1、患者3及患者4以聯合多種抗癲癇發作藥物控制癲癇發作,患者1發作間期VEEG大腦皮質雙側均有癇樣放電,臨床發作特點屬于頑固性局灶性發作,住院期間首先選用拉莫三嗪片無法控制右側肢體抽搐癥狀,后口服加用吡侖帕奈片也不能完全控制癥狀,仍有輕微右上肢抽搐癥狀,最后苯巴比妥注射液每12 h一次,完全控制癥狀。出院后停用苯巴比妥注射液,加用苯巴比妥片。患者2屬于難治性癲癇,1歲左右出現癲癇發作,期間反復調整抗癲癇藥物及聯合3~4種抗癲癇發作藥物仍不能控制癲癇發作,基本每天出現癲癇發作數次,最后予以外科手術治療行大腦半球離斷術,術后繼續口服奧卡西平片、左乙拉西坦片、拉考沙胺片等抗癲癇藥物治療。患者3發作間期VEEG癇樣放電局限于右側大腦半球皮質,入院前口服奧卡西平不能完全控制癲癇發作癥狀,后加用左乙拉西坦片可控制癲癇發作。患者4既往新生兒腦損傷、熱性驚厥等病史,本次發作間期VEEG癇樣放電局限于左側大腦半球皮質,首先使用左乙拉西坦不能控制癲癇發作,后加用奧卡西平控制癲癇發作。以上4例DDMS患者住院期間調整抗癲癇發作治療方案后,出院隨訪仍有間斷發作情況,總體而言癲癇發作控制較滿意。
DDMS是一種罕見的腦部綜合征,會導致癲癇發作。治療的主要目標是控制癲癇發作。為了提高疾病預后,癲癇專科醫生應該增強對DDMS的認識,提高該疾病的診斷率,并在早期發現和早期診斷方面做好工作,以便進行合理的治療。
利益沖突聲明 所有作者無利益沖突。