

引用本文: 劉愛杰, 許小菁, 孫丹, 曾琦, 楊小玲, 楊志仙, 吳曄, 劉曉燕, 姜玉武, 吳希如, 張月華. 原鈣黏蛋白19基因相關癲癇的遺傳特點及臨床表型譜研究. 癲癇雜志, 2017, 3(4): 283-291. doi: 10.7507/2096-0247.20170042 復制
原鈣黏蛋白19(X-linked protocadherin 19, PCDH19) 基因突變首先在限于女性的癲癇伴智力低下(Epilepsy and mental retardation limited to females,EFMR)家系中報道[1]。隨后研究發現PCDH19基因也是Dravet綜合征(Dravet syndrome,DS)的致病基因之一,而且少數PCDH19基因突變嵌合體的男性也可發病,說明該病已不再局限于女性[2-4]。此外有少數患者可不伴智力低下[5-7]。因此,目前該病多以PCDH19基因相關癲癇命名[8]。迄今國際上已有100多例女性患者和4例男性嵌合體患者報道,但國內有關該病報道尚少[9-12]。本文總結PCDH19基因相關癲癇患者的遺傳特點和臨床表型譜。
1 資料與方法
1.1 資料來源
本研究收集2005年2月-2017年4月在北京大學第一醫院兒科及武漢市兒童醫院神經內科確診的PCDH19基因相關癲癇41例先證者(其中38例來源于北京大學第一醫院兒科,3例來源于武漢市兒童醫院神經內科)。先證者中女39例,男2例。30例為散發患者,11例有癲癇和/或熱性驚厥家族史。對照組100例為無親緣關系的中國漢族健康成人,無神經系統疾病史。本研究通過北京大學第一醫院及武漢市兒童醫院倫理委員會的批準,所有患兒家長均簽署知情同意書。
入組標準:① 起病年齡1~60個月;② 癲癇發作類型包括強直發作、陣攣發作或強直陣攣發作、局灶性發作等;③ 發作具有叢集性的特點;④ PCDH19基因突變陽性。
1.2 研究方法
1.2.1 PCDH19基因突變檢測
采用PCR-Sanger測序或靶向捕獲二代測序(Next generation sequencing,NGS)的方法篩查PCDH19基因突變[11, 13, 14]。如測序未發現PCDH19基因突變,則進一步采用多重連接依賴的探針擴增技術(Multiplex ligation-dependent probe amplification,MLPA)方法篩查PCDH19基因的大片段缺失或重復[12]。對存在基因突變PCDH19基因突變的患兒家庭,進一步對患兒父母及其他可獲取的家系成員進行檢測。如發現未報道過的新突變,則對100例正常對照DNA進行該突變的檢測,以排除多態性。
1.2.2 臨床資料
總結先證者及家系受累成員的臨床表現;輔助檢查結果,包括腦電圖(EEG)、頭顱磁共振(MRI)等;以及抗癲癇藥物(AEDs)用藥史。
2 結果
2.1 PCDH19基因突變篩查結果
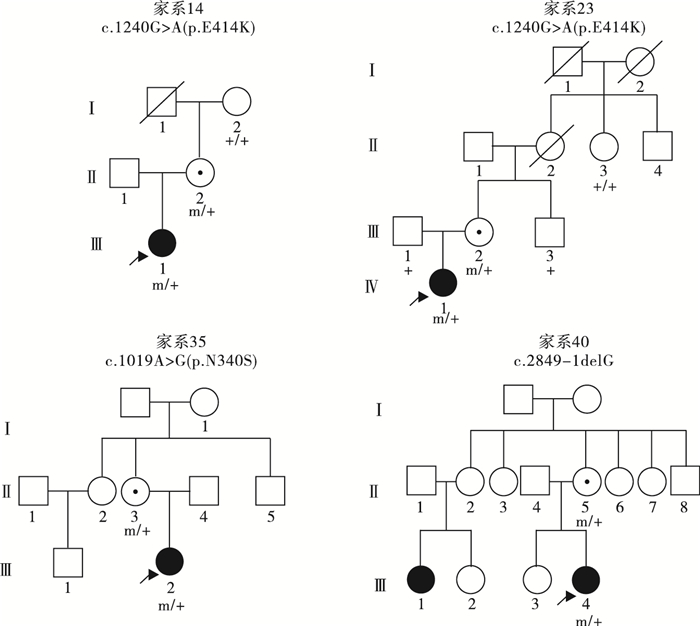
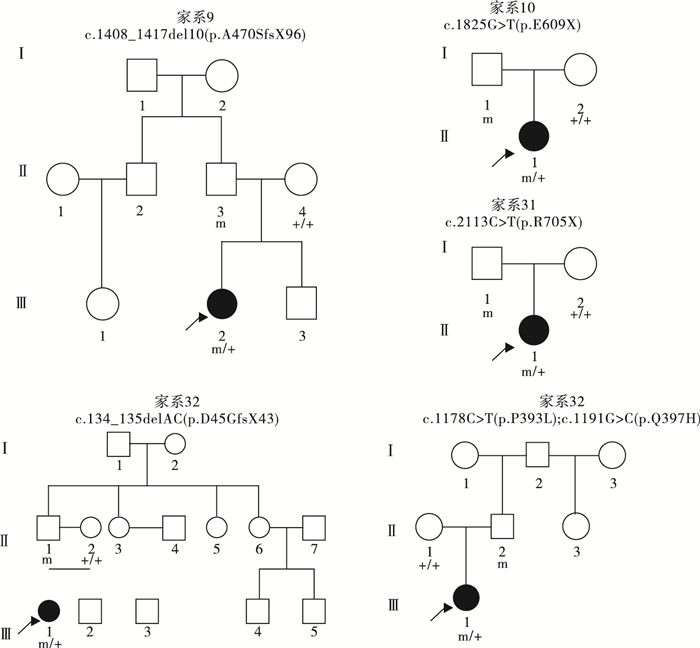
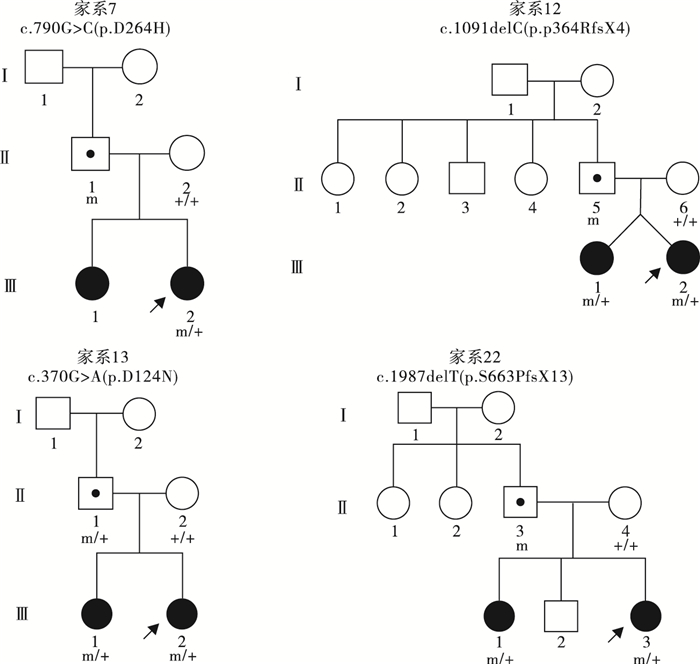
41例先證者中,通過測序發現40例(97.6%)攜帶PCDH19基因突變,MLPA發現1例(2.4%)為整個PCDH19基因缺失,見表 1。共檢測到37種突變(含12種國際未見報道的新突變),34種點突變位于胞外區的第1外顯子。12種新突變(表 1)在PCDH19基因突變數據庫、人類基因突變數據庫、外顯子組整合聯合數據庫和100例中國正常人群對照中均未發現。
2例男性患兒(先證者25和先證者26) 經NGS檢測發現為PCDH19基因突變嵌合體。先證者25為c.317T>A嵌合突變,突變等位基因占比為85%(73/85,85%)。先證者26為c.158dupT嵌合突變,其突變等位基因占比為33%(41/124,33%)。2例均經Sanger測序驗證為嵌合突變,且為新生突變。
2.2 女性PCDH19基因突變患者家系分析
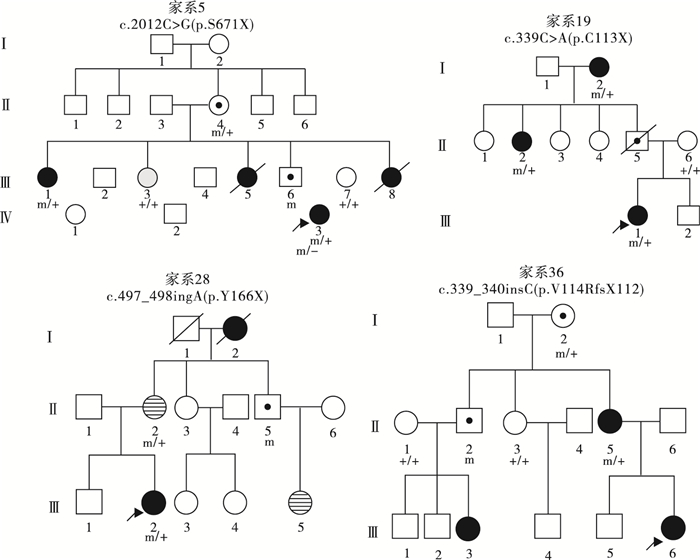
39例PCDH19基因突變陽性的女性患兒中,19例(19/39,48.7%)為遺傳性突變,20例(20/39,51.3%)為新生突變。19例攜帶遺傳性突變的先證者中,9例為父源遺傳,6例為母源遺傳,4例為家系遺傳,家系圖和PCDH19基因突變分析結果見圖 1~5。9例突變遺傳自父親的先證者中,5例為散發病例(先證者無同胞姐妹),4例有同胞姐妹且共患癲癇;其中1例先證者父親為PCDH19突變嵌合體(家系13),余8例先證者父親為半合子且父親的突變來源未知(因無法取得父親一方更多家系成員的血樣)。6例突變遺傳自母親的先證者中,3例為散發病例,2例母女共患癲癇,1例先證者大姨的女兒有癲癇;此6例患兒家系中,僅家系4取得完整的外祖父及外祖母血樣,證實母親為新生突變,且母親突變等位基因來自外祖父。4例三代遺傳的家系中,家系5和家系36先證者的祖母均為無癥狀突變攜帶者。
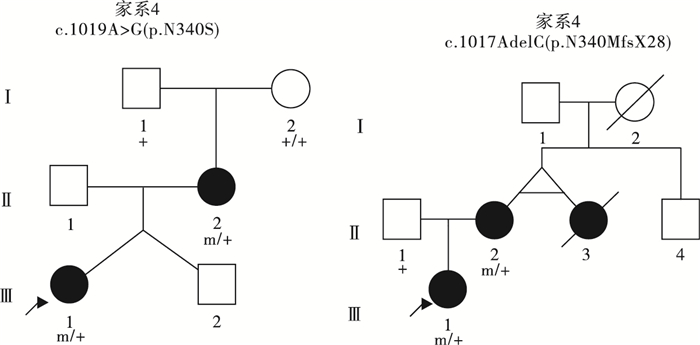
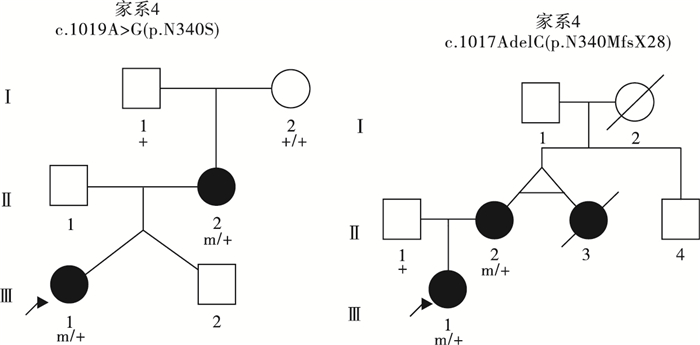 圖1
2例母女同患PCDH19基因相關癲癇的女性患兒家系圖
Figure1.
Two female PCDH19 gene related epilepsy pedigrees with affected mothers
圖1
2例母女同患PCDH19基因相關癲癇的女性患兒家系圖
Figure1.
Two female PCDH19 gene related epilepsy pedigrees with affected mothers
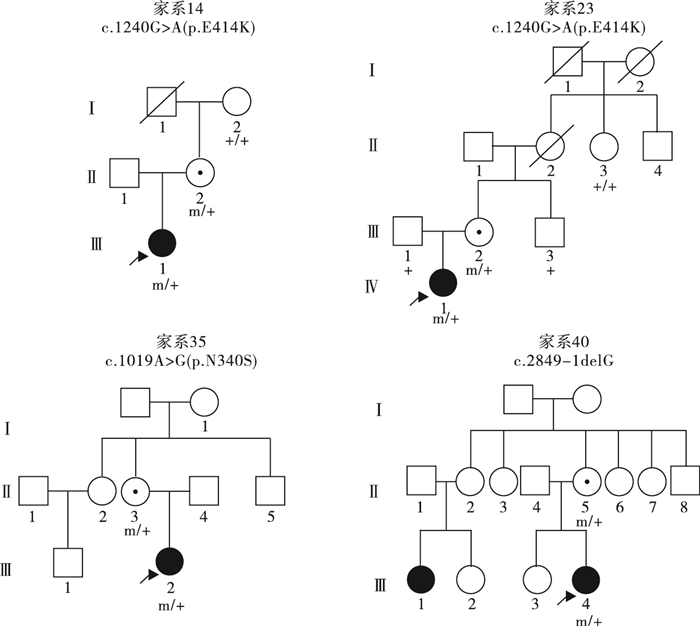 圖2
4例PCDH19基因突變遺傳自無癥狀母親的女性患兒家系圖
Figure2.
Four female PCDH19 gene related epilepsy pedigrees with asymptomatic mothers
圖2
4例PCDH19基因突變遺傳自無癥狀母親的女性患兒家系圖
Figure2.
Four female PCDH19 gene related epilepsy pedigrees with asymptomatic mothers
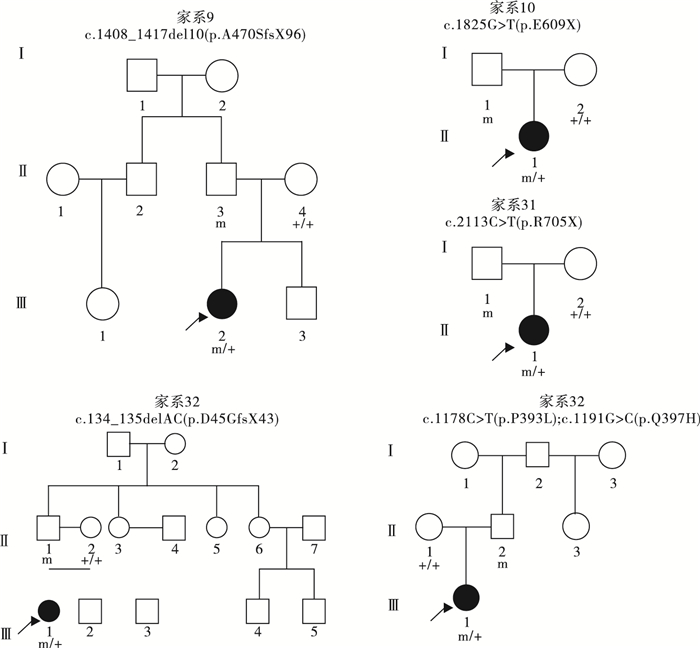 圖3
5例PCDH19基因突變遺傳自父親的散發女性患兒家系圖
Figure3.
Five sporadic female PCDH19 gene related epilepsy pedigrees with transmitting fathers
圖3
5例PCDH19基因突變遺傳自父親的散發女性患兒家系圖
Figure3.
Five sporadic female PCDH19 gene related epilepsy pedigrees with transmitting fathers
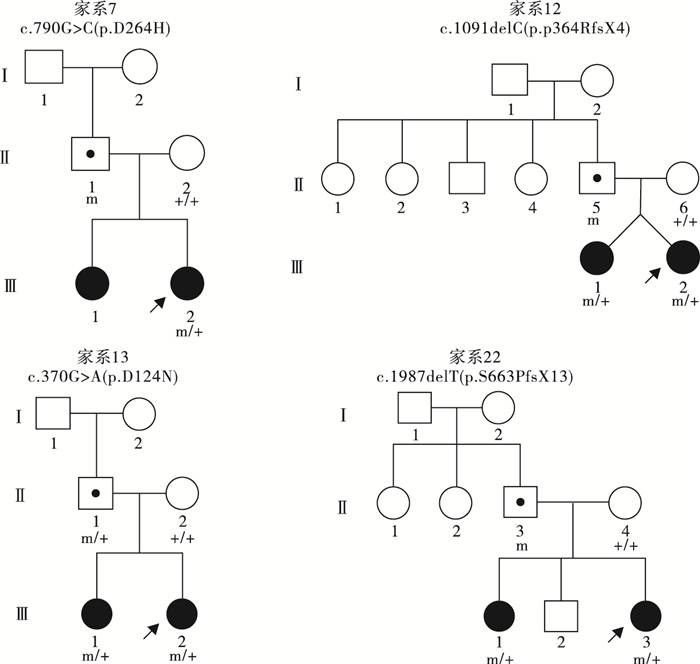 圖4
4例PCDH19基因突變遺傳自父親且姐妹共患的女性患兒家系圖
Figure4.
Four female PCDH19 gene related epilepsy pedigrees with transmitting fathers and affected sisters
圖4
4例PCDH19基因突變遺傳自父親且姐妹共患的女性患兒家系圖
Figure4.
Four female PCDH19 gene related epilepsy pedigrees with transmitting fathers and affected sisters
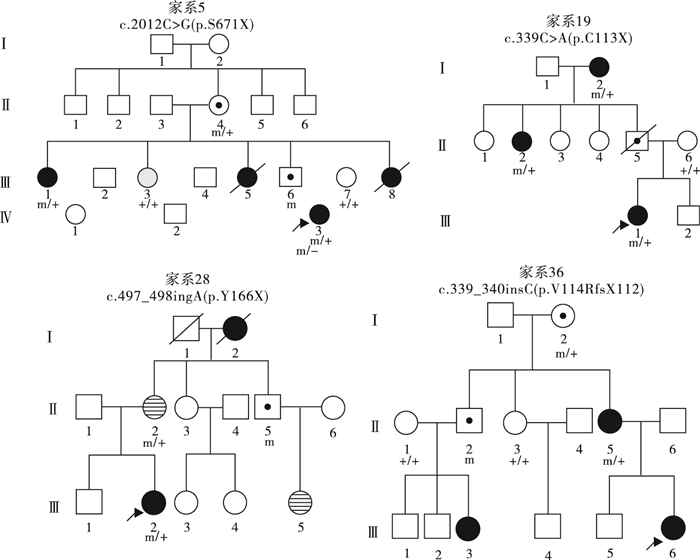 圖5
4例三代遺傳大家系的女性患兒家系圖
圖5
4例三代遺傳大家系的女性患兒家系圖
注:黑色表示癲癇患者,灰色表示有癲癇表型但無
2.3 PCDH19基因相關癲癇的臨床表型譜
本研究中的39例女性癲癇患者家系中,可以明確攜帶突變的女性59例,其中有臨床表現者53例,外顯率為90%。12例半合子父親和1例嵌合體父親均無臨床表型。攜帶PCDH19基因突變者的表型包括癲癇伴智力低下(40例,含7例Dravet綜合征),癲癇不伴智力低下(11例),熱性驚厥(2例),以及女性無癥狀攜帶者(6例)。即使在同一家系內,攜帶相同PCDH19基因突變的女性表現度各有不同。
總結41例先證者及5例家系受累成員(先證者12的異卵雙胎姐姐,先證者13的姐姐,先證者22的姐姐,先證者36的母親及舅舅的女兒)共46例患者的臨床特點。起病年齡4~42個月(中位起病年齡11個月),33例(33/46,71.7%)以全面強直陣攣發作(Generalized tonic clonic seizures, GTCSs)起病,13例(13/46,28.3%)以局灶性發作起病。
病程中發作類型包括GTCSs(40/46,87.0%),局灶性發作伴或不伴繼發GTCS(32/46,69.6%),肌陣攣發作(6/46,13.0%),不典型失神(3/46,6.5%)和失張力發作(1/46,2.2%),其中8例患者有多種發作類型。叢集性發作見于所有患者(46/46,100%),80.4%(37/46) 患者發作具有熱敏感的特點,僅3例曾有發熱誘發的癲癇持續狀態。35例(35/46,76%)患者有不同程度的智力損害,7例(7/46,15.2%)有孤獨癥樣表現。
46例患者中,45例進行過至少一次EEG檢查。15例有背景活動慢波;21例發作間期監測到局灶性癲癇放電;20例監測到癲癇發作,其中13例監測到局灶性或多灶性發作,6例監測到GTCSs或陣攣-強直-陣攣發作),1例(先證者17) 監測到多種發作類型,包括局灶性發作、肌陣攣發作和GTCSs。41例先證者行頭顱MRI檢查,均未見明顯異常。
46例患者中,40例對AEDs的療效欠佳,均嘗試多種AEDs治療。所用AEDs包括丙戊酸(VPA)、托吡酯(TPM)、左乙拉西坦(LEV)、氯硝西泮(CZP)、硝西泮(NZP)、卡馬西平(CBZ)、奧卡西平(OXC)、拉莫三嗪(LTG)和苯巴比妥(PB)。末次隨訪(患者年齡1~42歲)時12例單藥治療(VPA、LEV或TPM),22例用兩種AEDs,10例用三種AEDs治療,2例(先證者25和先證者36的母親)未規律服藥。30例發作頻率減少,發作緩解間隔為4個月~7年,其中1年以上無發作者9例,發作緩解時間最長者已7年無發作。1年以上無發作者服用的AEDs有VPA,TPM,LEV,PB,CZP和LTG。病程中發作間隔曾經>1年,之后再次出現發作者11例,其中3例因發熱誘發;10例曾在減藥過程中或停藥后再次發作。
3 討論
PCDH19基因相關癲癇為特殊的X連鎖遺傳,即女性雜合子受累,男性半合子不受累[1]。“細胞干擾”機制是解釋這種特殊遺傳方式的主要假說,該假說推測當個體表達兩種不同PCDH19蛋白時,細胞與細胞之間正常的相互作用會被干擾,從而發病[2]。男性嵌合體患者的發現似乎進一步證實了該假說[2-4]。
雖然PCDH19基因突變最早是在EFMR家系中發現,但隨后的研究證實PCDH19基因突變導致的癲癇散發病例更多,且多為新生突變[15, 16]。本研究41例先證者中,30例為散發病例,僅11例有家族史。新生突變和遺傳性突變比例接近,均占50%,散發病例的遺傳性突變可來源于無癥狀的父親或母親。總結本研究和所有文獻報道的226例患者的PCDH19基因致病突變,共發現162種不同的突變,包括各種突變類型,其中46.9%(76/162) 為提前終止密碼子突變,這種突變通常會因無義介導的mRNA降解機制而導致蛋白功能缺失[1]。迄今為止,所有致病的錯義突變均位于第1外顯子的胞外區,推測此類突變可能會影響蛋白的黏附功能[17]。本研究發現的37種突變,14種為國際已報道的突變,其余23種為本課題組在中國人群發現。
到目前為止,國際上僅有4例男性嵌合體發病的報道,1例為整個PCDH19基因缺失,3例為點突變。本研究在中國人群中發現3例男性PCDH19基因點突變嵌合體,2例為癲癇患者,1例為無癥狀父親。男性無癥狀嵌合體的發現對既往的“細胞干擾”機制假說是一個挑戰,因此我們提出男性PCDH19基因突變嵌合體的表型與其體內突變型和野生型細胞的比例也可能有關。
PCDH19基因相關癲癇具有外顯率不全和表型異質性特點[1, 18-21]。本研究在41個PCDH19基因相關癲癇患者的家系中,發現攜帶PCDH19基因突變者的表型包括癲癇伴智力低下(含Dravet綜合征)、癲癇不伴智力低下、熱性驚厥和女性無癥狀攜帶者。本組攜帶突變的女性外顯率為90%,低于Dibbens等報道的97%的外顯率[1]。該病具體的外顯率尚需要更多的病例進行驗證。本組病例的表型異質性明顯,同一突變在不同個體,甚至是攜帶相同突變的同一家系成員之間的表現度也具有較大差異。本病以GTCSs和局灶性發作為主要發作類型,叢集性發作和發作具有熱敏感性為其主要的臨床特點[15]。
綜上所述,PCDH19基因突變可為遺傳性突變或新生突變,受累者以女性為主,少數男性嵌合體可發病或無癥狀。PCDH19基因相關癲癇具有外顯率不全和表型異質性,以GTCSs和局灶性發作為主、叢集性發作和熱敏感為其主要臨床特點。隨著對PCDH19基因相關癲癇的臨床和遺傳學特點認識的不斷提高,越來越多的患兒將得到明確診斷和合理治療,并可為家庭的遺傳咨詢提供指導。
原鈣黏蛋白19(X-linked protocadherin 19, PCDH19) 基因突變首先在限于女性的癲癇伴智力低下(Epilepsy and mental retardation limited to females,EFMR)家系中報道[1]。隨后研究發現PCDH19基因也是Dravet綜合征(Dravet syndrome,DS)的致病基因之一,而且少數PCDH19基因突變嵌合體的男性也可發病,說明該病已不再局限于女性[2-4]。此外有少數患者可不伴智力低下[5-7]。因此,目前該病多以PCDH19基因相關癲癇命名[8]。迄今國際上已有100多例女性患者和4例男性嵌合體患者報道,但國內有關該病報道尚少[9-12]。本文總結PCDH19基因相關癲癇患者的遺傳特點和臨床表型譜。
1 資料與方法
1.1 資料來源
本研究收集2005年2月-2017年4月在北京大學第一醫院兒科及武漢市兒童醫院神經內科確診的PCDH19基因相關癲癇41例先證者(其中38例來源于北京大學第一醫院兒科,3例來源于武漢市兒童醫院神經內科)。先證者中女39例,男2例。30例為散發患者,11例有癲癇和/或熱性驚厥家族史。對照組100例為無親緣關系的中國漢族健康成人,無神經系統疾病史。本研究通過北京大學第一醫院及武漢市兒童醫院倫理委員會的批準,所有患兒家長均簽署知情同意書。
入組標準:① 起病年齡1~60個月;② 癲癇發作類型包括強直發作、陣攣發作或強直陣攣發作、局灶性發作等;③ 發作具有叢集性的特點;④ PCDH19基因突變陽性。
1.2 研究方法
1.2.1 PCDH19基因突變檢測
采用PCR-Sanger測序或靶向捕獲二代測序(Next generation sequencing,NGS)的方法篩查PCDH19基因突變[11, 13, 14]。如測序未發現PCDH19基因突變,則進一步采用多重連接依賴的探針擴增技術(Multiplex ligation-dependent probe amplification,MLPA)方法篩查PCDH19基因的大片段缺失或重復[12]。對存在基因突變PCDH19基因突變的患兒家庭,進一步對患兒父母及其他可獲取的家系成員進行檢測。如發現未報道過的新突變,則對100例正常對照DNA進行該突變的檢測,以排除多態性。
1.2.2 臨床資料
總結先證者及家系受累成員的臨床表現;輔助檢查結果,包括腦電圖(EEG)、頭顱磁共振(MRI)等;以及抗癲癇藥物(AEDs)用藥史。
2 結果
2.1 PCDH19基因突變篩查結果
41例先證者中,通過測序發現40例(97.6%)攜帶PCDH19基因突變,MLPA發現1例(2.4%)為整個PCDH19基因缺失,見表 1。共檢測到37種突變(含12種國際未見報道的新突變),34種點突變位于胞外區的第1外顯子。12種新突變(表 1)在PCDH19基因突變數據庫、人類基因突變數據庫、外顯子組整合聯合數據庫和100例中國正常人群對照中均未發現。
2例男性患兒(先證者25和先證者26) 經NGS檢測發現為PCDH19基因突變嵌合體。先證者25為c.317T>A嵌合突變,突變等位基因占比為85%(73/85,85%)。先證者26為c.158dupT嵌合突變,其突變等位基因占比為33%(41/124,33%)。2例均經Sanger測序驗證為嵌合突變,且為新生突變。
2.2 女性PCDH19基因突變患者家系分析
39例PCDH19基因突變陽性的女性患兒中,19例(19/39,48.7%)為遺傳性突變,20例(20/39,51.3%)為新生突變。19例攜帶遺傳性突變的先證者中,9例為父源遺傳,6例為母源遺傳,4例為家系遺傳,家系圖和PCDH19基因突變分析結果見圖 1~5。9例突變遺傳自父親的先證者中,5例為散發病例(先證者無同胞姐妹),4例有同胞姐妹且共患癲癇;其中1例先證者父親為PCDH19突變嵌合體(家系13),余8例先證者父親為半合子且父親的突變來源未知(因無法取得父親一方更多家系成員的血樣)。6例突變遺傳自母親的先證者中,3例為散發病例,2例母女共患癲癇,1例先證者大姨的女兒有癲癇;此6例患兒家系中,僅家系4取得完整的外祖父及外祖母血樣,證實母親為新生突變,且母親突變等位基因來自外祖父。4例三代遺傳的家系中,家系5和家系36先證者的祖母均為無癥狀突變攜帶者。
圖1
2例母女同患PCDH19基因相關癲癇的女性患兒家系圖
Figure1.
Two female PCDH19 gene related epilepsy pedigrees with affected mothers
圖2
4例PCDH19基因突變遺傳自無癥狀母親的女性患兒家系圖
Figure2.
Four female PCDH19 gene related epilepsy pedigrees with asymptomatic mothers
圖3
5例PCDH19基因突變遺傳自父親的散發女性患兒家系圖
Figure3.
Five sporadic female PCDH19 gene related epilepsy pedigrees with transmitting fathers
圖4
4例PCDH19基因突變遺傳自父親且姐妹共患的女性患兒家系圖
Figure4.
Four female PCDH19 gene related epilepsy pedigrees with transmitting fathers and affected sisters
圖5
4例三代遺傳大家系的女性患兒家系圖
注:黑色表示癲癇患者,灰色表示有癲癇表型但無
2.3 PCDH19基因相關癲癇的臨床表型譜
本研究中的39例女性癲癇患者家系中,可以明確攜帶突變的女性59例,其中有臨床表現者53例,外顯率為90%。12例半合子父親和1例嵌合體父親均無臨床表型。攜帶PCDH19基因突變者的表型包括癲癇伴智力低下(40例,含7例Dravet綜合征),癲癇不伴智力低下(11例),熱性驚厥(2例),以及女性無癥狀攜帶者(6例)。即使在同一家系內,攜帶相同PCDH19基因突變的女性表現度各有不同。
總結41例先證者及5例家系受累成員(先證者12的異卵雙胎姐姐,先證者13的姐姐,先證者22的姐姐,先證者36的母親及舅舅的女兒)共46例患者的臨床特點。起病年齡4~42個月(中位起病年齡11個月),33例(33/46,71.7%)以全面強直陣攣發作(Generalized tonic clonic seizures, GTCSs)起病,13例(13/46,28.3%)以局灶性發作起病。
病程中發作類型包括GTCSs(40/46,87.0%),局灶性發作伴或不伴繼發GTCS(32/46,69.6%),肌陣攣發作(6/46,13.0%),不典型失神(3/46,6.5%)和失張力發作(1/46,2.2%),其中8例患者有多種發作類型。叢集性發作見于所有患者(46/46,100%),80.4%(37/46) 患者發作具有熱敏感的特點,僅3例曾有發熱誘發的癲癇持續狀態。35例(35/46,76%)患者有不同程度的智力損害,7例(7/46,15.2%)有孤獨癥樣表現。
46例患者中,45例進行過至少一次EEG檢查。15例有背景活動慢波;21例發作間期監測到局灶性癲癇放電;20例監測到癲癇發作,其中13例監測到局灶性或多灶性發作,6例監測到GTCSs或陣攣-強直-陣攣發作),1例(先證者17) 監測到多種發作類型,包括局灶性發作、肌陣攣發作和GTCSs。41例先證者行頭顱MRI檢查,均未見明顯異常。
46例患者中,40例對AEDs的療效欠佳,均嘗試多種AEDs治療。所用AEDs包括丙戊酸(VPA)、托吡酯(TPM)、左乙拉西坦(LEV)、氯硝西泮(CZP)、硝西泮(NZP)、卡馬西平(CBZ)、奧卡西平(OXC)、拉莫三嗪(LTG)和苯巴比妥(PB)。末次隨訪(患者年齡1~42歲)時12例單藥治療(VPA、LEV或TPM),22例用兩種AEDs,10例用三種AEDs治療,2例(先證者25和先證者36的母親)未規律服藥。30例發作頻率減少,發作緩解間隔為4個月~7年,其中1年以上無發作者9例,發作緩解時間最長者已7年無發作。1年以上無發作者服用的AEDs有VPA,TPM,LEV,PB,CZP和LTG。病程中發作間隔曾經>1年,之后再次出現發作者11例,其中3例因發熱誘發;10例曾在減藥過程中或停藥后再次發作。
3 討論
PCDH19基因相關癲癇為特殊的X連鎖遺傳,即女性雜合子受累,男性半合子不受累[1]。“細胞干擾”機制是解釋這種特殊遺傳方式的主要假說,該假說推測當個體表達兩種不同PCDH19蛋白時,細胞與細胞之間正常的相互作用會被干擾,從而發病[2]。男性嵌合體患者的發現似乎進一步證實了該假說[2-4]。
雖然PCDH19基因突變最早是在EFMR家系中發現,但隨后的研究證實PCDH19基因突變導致的癲癇散發病例更多,且多為新生突變[15, 16]。本研究41例先證者中,30例為散發病例,僅11例有家族史。新生突變和遺傳性突變比例接近,均占50%,散發病例的遺傳性突變可來源于無癥狀的父親或母親。總結本研究和所有文獻報道的226例患者的PCDH19基因致病突變,共發現162種不同的突變,包括各種突變類型,其中46.9%(76/162) 為提前終止密碼子突變,這種突變通常會因無義介導的mRNA降解機制而導致蛋白功能缺失[1]。迄今為止,所有致病的錯義突變均位于第1外顯子的胞外區,推測此類突變可能會影響蛋白的黏附功能[17]。本研究發現的37種突變,14種為國際已報道的突變,其余23種為本課題組在中國人群發現。
到目前為止,國際上僅有4例男性嵌合體發病的報道,1例為整個PCDH19基因缺失,3例為點突變。本研究在中國人群中發現3例男性PCDH19基因點突變嵌合體,2例為癲癇患者,1例為無癥狀父親。男性無癥狀嵌合體的發現對既往的“細胞干擾”機制假說是一個挑戰,因此我們提出男性PCDH19基因突變嵌合體的表型與其體內突變型和野生型細胞的比例也可能有關。
PCDH19基因相關癲癇具有外顯率不全和表型異質性特點[1, 18-21]。本研究在41個PCDH19基因相關癲癇患者的家系中,發現攜帶PCDH19基因突變者的表型包括癲癇伴智力低下(含Dravet綜合征)、癲癇不伴智力低下、熱性驚厥和女性無癥狀攜帶者。本組攜帶突變的女性外顯率為90%,低于Dibbens等報道的97%的外顯率[1]。該病具體的外顯率尚需要更多的病例進行驗證。本組病例的表型異質性明顯,同一突變在不同個體,甚至是攜帶相同突變的同一家系成員之間的表現度也具有較大差異。本病以GTCSs和局灶性發作為主要發作類型,叢集性發作和發作具有熱敏感性為其主要的臨床特點[15]。
綜上所述,PCDH19基因突變可為遺傳性突變或新生突變,受累者以女性為主,少數男性嵌合體可發病或無癥狀。PCDH19基因相關癲癇具有外顯率不全和表型異質性,以GTCSs和局灶性發作為主、叢集性發作和熱敏感為其主要臨床特點。隨著對PCDH19基因相關癲癇的臨床和遺傳學特點認識的不斷提高,越來越多的患兒將得到明確診斷和合理治療,并可為家庭的遺傳咨詢提供指導。